天然产物 (−)-Caulamidine A (1)的全合成
天然产物三卤化吲哚类生物碱Caulamidine A(1)和B(2)是由美国科学家Gustafson及其同事于2004年从海洋苔藓虫Caulibugula intermis的提取物中作为次要成分分离获得 (图1)[1],并在微摩尔浓度下对氯喹敏感和耐药恶性疟原虫菌株表现出良好的生物活性,且对人体细胞几乎没有毒性。
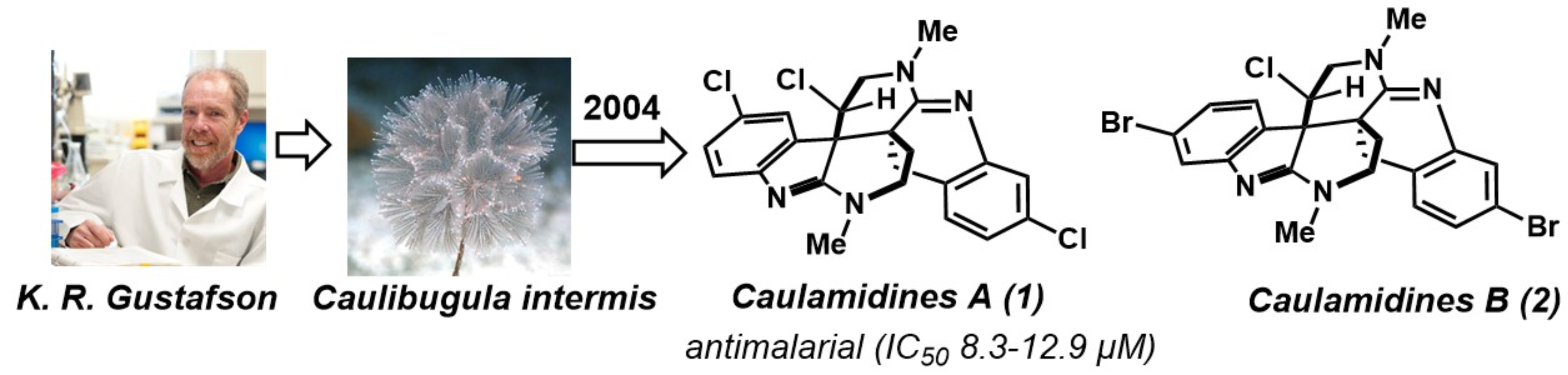
图1 Caulamidine A(1)的分离
由于其丰度较低和独特的结构,直到2018年科学家借助现代核磁共振技术和计算化学才完成对Caulamidine A(1)和B(2)相对立体化学的解析[2],即结构上,(1)和(2)均具有六氢-2,6-萘啶以及二氢吲哚衍生的四氢喹啉相互稠合的环系统(图 2)。虽然科学家对其进行了许多研究,但对其生物合成途径以及全合成尚待研究。
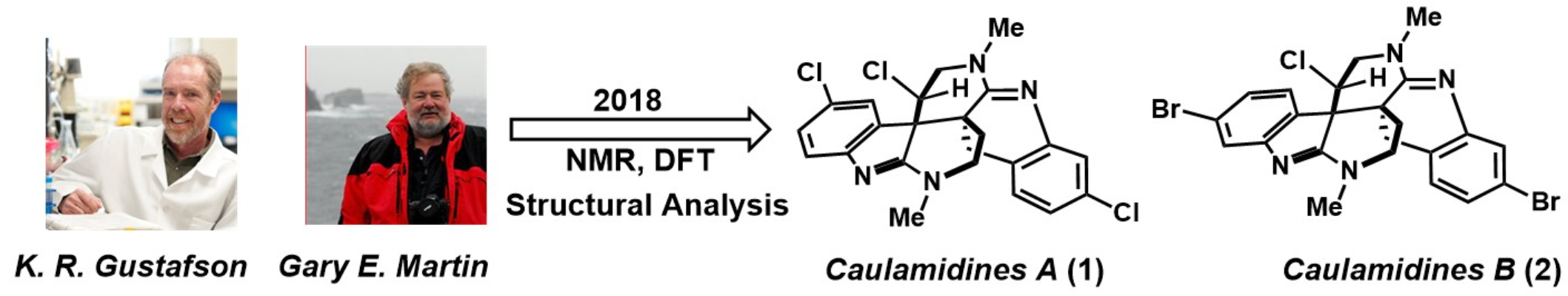
图 2 Caulamidine A(1)和(2)的结构解析
近日,美国University of California−Berkeley的Thomas J. Maimone团队在J. Am. Chem. Soc上,以 “Enantioselective Total Synthesis of (−)-Caulamidine A” 为题,首次报道了天然产物(−)-Caulamidine A(1)全合成路线设计,并确定了(−)-Caulamidine A的绝对构型。
其全合成步骤如下
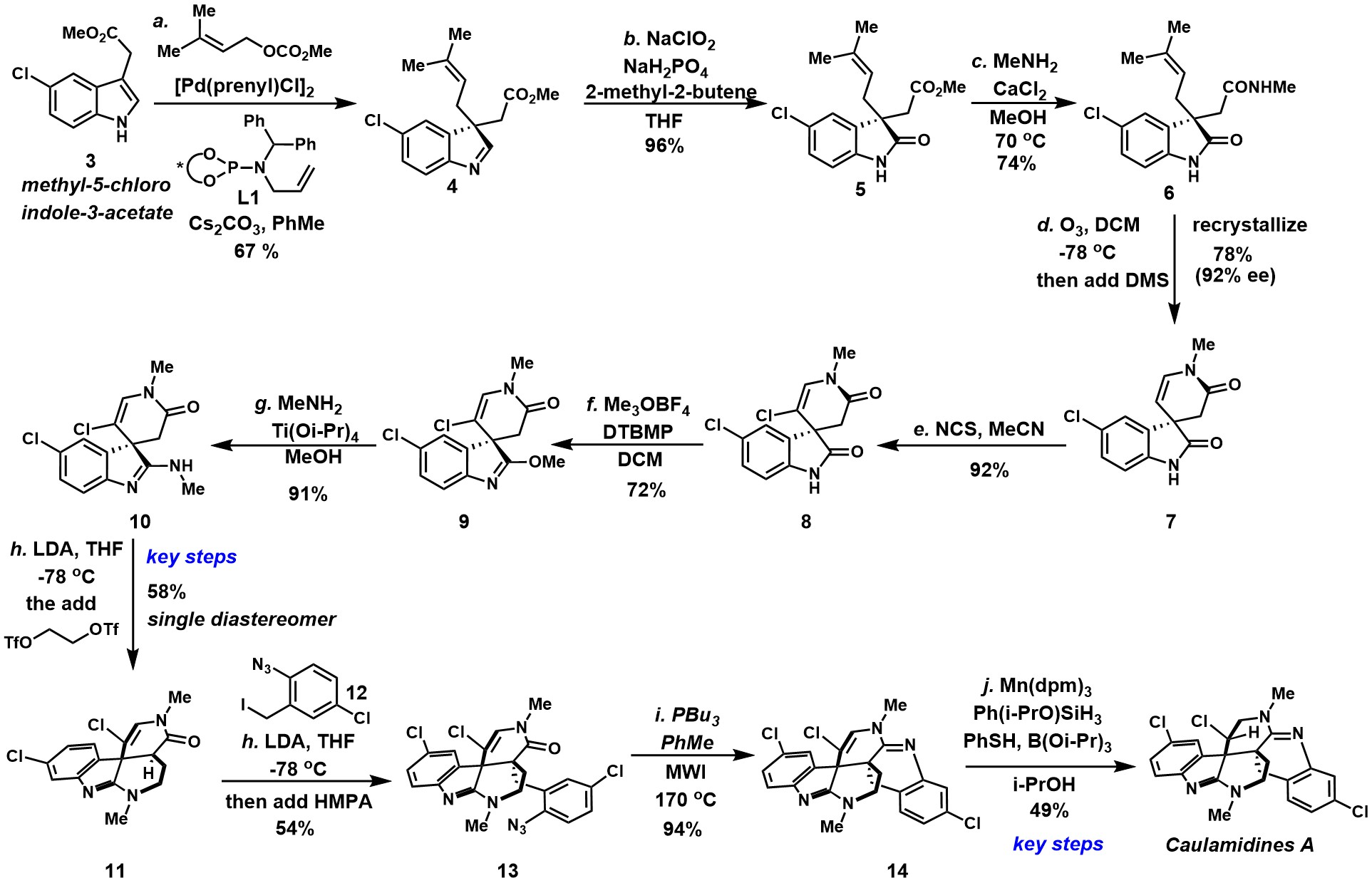
(图片来源:J. Am. Chem. Soc.)
以甲基-5-氯吲哚-3-乙酸酯(3)为原料,通过You 's pd催化的不对称异戊烯化反应获得了单一构型3-异戊烯基吲哚啉(4)[3],而后(4)通过Lindgren-Kraus氧化获得氧化吲哚(5)。(5)在氯化钙的条件下将酯转化为酰胺(6)[4],随后(6)的烯烃骨架发生臭氧分解以及与仲酰胺的原位缩合获得砌块螺环(7)(并通过重结晶提高其对映体纯度92%),而后(7)发生NBS溴代生成(8)。其次,(8)在Meerwein 's盐和2,6-二叔丁基-4-甲基吡啶(DTBMP)发生O-烷基化反应获得砌块(9),(9)在甲胺和Ti(Oi-Pr)4条件下获得环化前体(10)。双三氟酸乙二醇(处理(10)可以58%的产率获得关键的砌块四环(11)。 (12)经烯醇化、以及与(12)SN2反应获得了单一非对映体的叠氮化物(13), 而后微波辐照下的发生Staudinger-aza-Wittig反应(PBu3)生成双脒(14),顺利完成六环核心骨架的构建。最后,(14)通过Mn介导的高度非对映选择性氢原子转移构建关键的含氯的立体中心,从而可以49%的分离产率生成(−)-caulamidine A(1)。(1的光谱和分析数据与文献报道的值非常一致)。
Thomas J. Maimone教授以甲基-5-氯吲哚-3-乙酸酯为起始原料,以11步最长线性步骤首次完成了天然产物(−)-Caulamidine A (1)全合成路线设计,并确定了(−)-Caulamidine A(1)的绝对构型。 其中的关键步骤主要涉及glycol bistriflat促进非对映选择性酮-脒环化反应;高度非对映选择性氢原子转移构建关键的含氯的立体中心。
原文:Enantioselective Total Synthesis of (−)-Caulamidine A.
Zhouyang Zhu and Thomas J. Maimone*
J. Am. Chem. Soc., 2023, https://doi.org/10.1021/jacs.3c04493
参考文献
- D. J. Milanowski, K. R. Gustafson, J. A. Kelley, J. B. McMahon, J. Nat. Prod. 2004, 67, 70-73. DOI: 10.1021/np030378l
- D. J. Milanowski, N. Oku, L. K. Cartner, H. R. Bokesch, R. T. Williamson, J. Saurí, Y. Liu, K. A. Blinov, Y. Ding, X.-C. Li, D. Ferreira, L. A. Walker, S. Khan, M. T. Davies-Coleman, J. A. Kelley, J. B. McMahon, G. E. Martin, K. R. Gustafson, Chem. Sci. 2018, 9, 307-314. doi: DOI:10.1039/C7SC01996C.
- H.-F. Tu, X. Zhang, C. Zheng, M. Zhu, S.-L. You, Nat. Catal. 2018, 1, 601-608. DOI: 10.1038/s41929-018-0111-8.
- M. W. Bundesmann, S. B. Coffey, S. W. Wright, Tetrahedron Lett. 2010, 51, 3879-3882. DOI: 10.1016/j.tetlet.2010.05.075.
相关化合物
| 品名 | CAS | 货号 |
|---|---|---|
| (R)-(+)-1,1'-Bi-2,2'-naphthol, 99% (R)-(+)-1,1'-联萘-2,2'-二酚 , 99% | 18531-94-7 | 465317 |
| 2-Methyl-2-butene 2-甲基-2-丁烯 , 95.0%(GC) | 513-35-9 | P0067 |
| Tris[(2,2,6,6-tetramethyl-5-oxo-3-hepten-3-yl)oxy]manganese 三[(2,2,6,6-四甲基-5-氧代-3-庚烯-3-基)氧基]锰 , ≥97% | 14324-99-3 | SY072824 |
相关阅读
七大系列,上万个产品,助力天然产物全合成研究!天然产物化学——助力提取、修饰、全合成、药理与代谢研究
光延反应的关键中间体——DEAD/DIAD,用于天然产物全合成
服务科技与工业发展 造福人类

关注微信公众号

