天然产物(+)-Ineleganolide的全合成
呋喃丁烯内酯(furanobutenolide)衍生的二萜(+)-Ineleganolide(1)是由中国台湾的化学家Duh及其同事于1999年从台湾的Formosan软珊瑚Sinularia inelegans中分离获得的一种对P-380白血病细胞系具有初步细胞毒性的物质(图1)[1]。虽然Vanderwal等人做出了努力[2],但由于 Ineleganolide 独特而具有挑战性的框架,在过去的二十年里, 其合成难题一直尚未解决。

图 1天然产物(+)-Ineleganolide的分离
(图片来源于J. Am. Chem. Soc)
天然产物(+)-Ineleganolide(1)具有特殊的骨架(图 1),即其含有一个高度刚性的氧化骨架,该骨架含有一个关键的中心七元环、一个远端异丙烯基和一个桥接的β-酮基四氢呋喃单元。
基于其特殊的骨架,目前仅有贝勒大学的John L. Wood教授[3]和加州理工学院Brian M. Stoltz教授分别以20步和23步线性最长步骤完成了二萜(+)-Ineleganolide的全合成。

Brian M. Stoltz教授开发的合成路线如下,
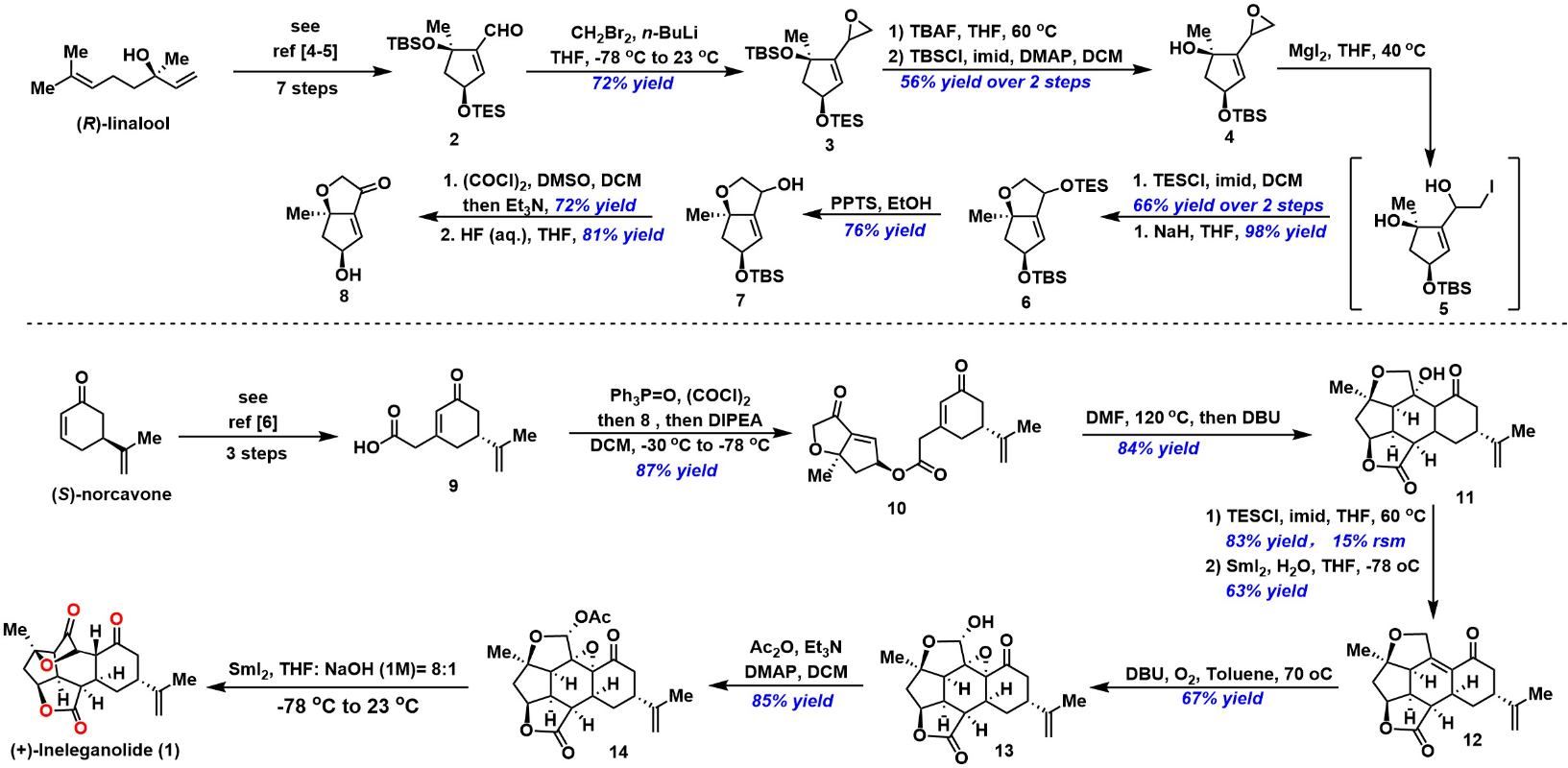
图3天然产物(+)-Ineleganolide的全合成
(图片来源于J. Am. Chem. Soc)
首先,作者以(R)-芳樟醇为原料[4-5],经7步反应获得醛砌块2。2经过环氧化反应获得环氧砌块3,3经脱除硅基保护、TBSCl可选择性对二级醇进行硅基化获得三级醇砌块4。中间体4发生开环反应、TBSCl对二级醇进行选择性的硅基化反应以及分子内取代反应三步获得双环中间体6。双环中间体6在PPTS/EtOH条件下进行选择性的脱保护获得醇中间体7,7发生Swern氧化反应和脱保护两步获得烯酮砌块8。
其次,以(S)-去甲香芹酮为原料[6],经三步反应获得羧酸砌块9,9与8在Ph3P=O/(COCl)2/DIPEA条件下进行酯化反应获得中间体10。10在DMF溶剂中进行高温加热,随后加入DBU进行分子内Michael加成反应获得五环中间体11。然后,11经三级醇进行选择性的硅基化反应和Sm介导的还原消除合成中间体12。12在DBU/O2条件下进行氧化获得环氧中间体13。
最后,环氧化物半缩醛13发生乙酰化合成中间体14,14经Semipinacol重排[7]完成 (+)-Ineleganolide(1)的全合成。
Brian M. Stoltz教授团队呋喃丁烯内酯(furanobutenolide)衍生的二萜(+)-Ineleganolide的全合成路线关键步骤主要涉及两个对映富集砌块的偶联(the coupling of two enantioenriched fragments); Michael加成和Aldol串联反应(Michael addition and aldol cascade); O2促进的C–H氧化(O2-facilitated C–H oxidation); 二碘化钐诱导的半频哪醇重排(samarium diiodide-induced semipinacol rearrangement)。
原文:“A Convergent Total Synthesis of (+)-Ineleganolide Benjamin M. Gross, Seo-Jung Han, Scott C. Virgil, and Brian M. Stoltz* J. Am. Chem. Soc., 2023, 145, 7763-7767. https://pubs.acs.org/doi/10.1021/jacs.3c02142
参考文献
- C.-Y. Duh, S.-K. Wang, M.-C. Chia, M. Y. Chiang, Tetrahedron Lett. 1999, 40, 6033. doi: 10.1016/S0040-4039(99)01194-6.
- E. J. Horn, J. S. Silverston, C. D. Vanderwal, J. Org. Chem. 2016, 81, 1819. doi: 10.1021/acs.joc.5b02550.
- J. P. Tuccinardi, J. L. Wood, J. Am. Chem. Soc. 2022, 144, 20539. doi : 10.1021/jacs.2c09826.
- Z. G. Brill, H. K. Grover, T. J. Maimone, 2016, 352, 1078. doi: 10.1126/science.aaf6742.
- N. J. Hafeman, M. Chan, T. J. Fulton, E. J. Alexy, S. A. Loskot, S. C. Virgil, B. M. Stoltz, J. Am. Chem. Soc. 2022, 144, 20232. doi: 10.1021/jacs.2c09583.
- I. I. R. A. Craig, J. L. Roizen, R. C. Smith, A. C. Jones, S. C. Virgil, B. M. Stoltz, Chem. Sci. 2017, 8, 507. doi: 10.1039/C6SC03347D.
- Z.-L. Song, C.-A. Fan, Y.-Q. Tu, Chem. Rev. 2011, 111, 7523. doi: 10.1021/cr200055g.
相关化合物
| 品名 | CAS | 货号 |
|---|---|---|
| (R)-(-)-Linalool, 95% (sum of enantiomers) (R)-(-)-Linalool, 95% (sum of enantiomers) | 126-91-0 | C42831 |
| tert-Butyldimethylchlorosilane, 99% tert-Butyldimethylchlorosilane, 99% | 18162-48-6 | 236144 |
| Ethyl acetate, 99.8%, for biochemistry, J&KSeal 乙酸乙酯 , 99.8% , 用于生物化学, J&KSeal瓶 | 141-78-6 | 925296 |
| N,N-Dimethylformamide, 99.9%, for synthesis N,N-二甲基甲酰胺 , 99.9% , 用于合成 | 68-12-2 | 914616 |
| N,N-Diisopropylethylamine, 99.5% N,N-二异丙基乙胺 , 99.5% | 7087-68-5 | 203402 |
相关阅读
七大系列,上万个产品,助力天然产物全合成研究!天然产物化学——助力提取、修饰、全合成、药理与代谢研究
光延反应的关键中间体——DEAD/DIAD,用于天然产物全合成
服务科技与工业发展 造福人类

关注微信公众号

